CompLife2007, Utrecht/NL. Day 1 and 2
CompLife 2007 was held 1.5 weeks ago in Utrecht, The Netherlands. The number of participants was much lower than last year in Cambridge. Ola and I gave a tutorial on Bioclipse, and Thorsten one on KNIME. Since a visit to Konstance to meet the KNIME developers, I had not been able to develop a KNIME plugin, but this was a nice opportunity to finally do so. I managed to do so, and wrote up a plugin that takes InChIKeys and then goes of the ChemSpider to download MDL molfiles:
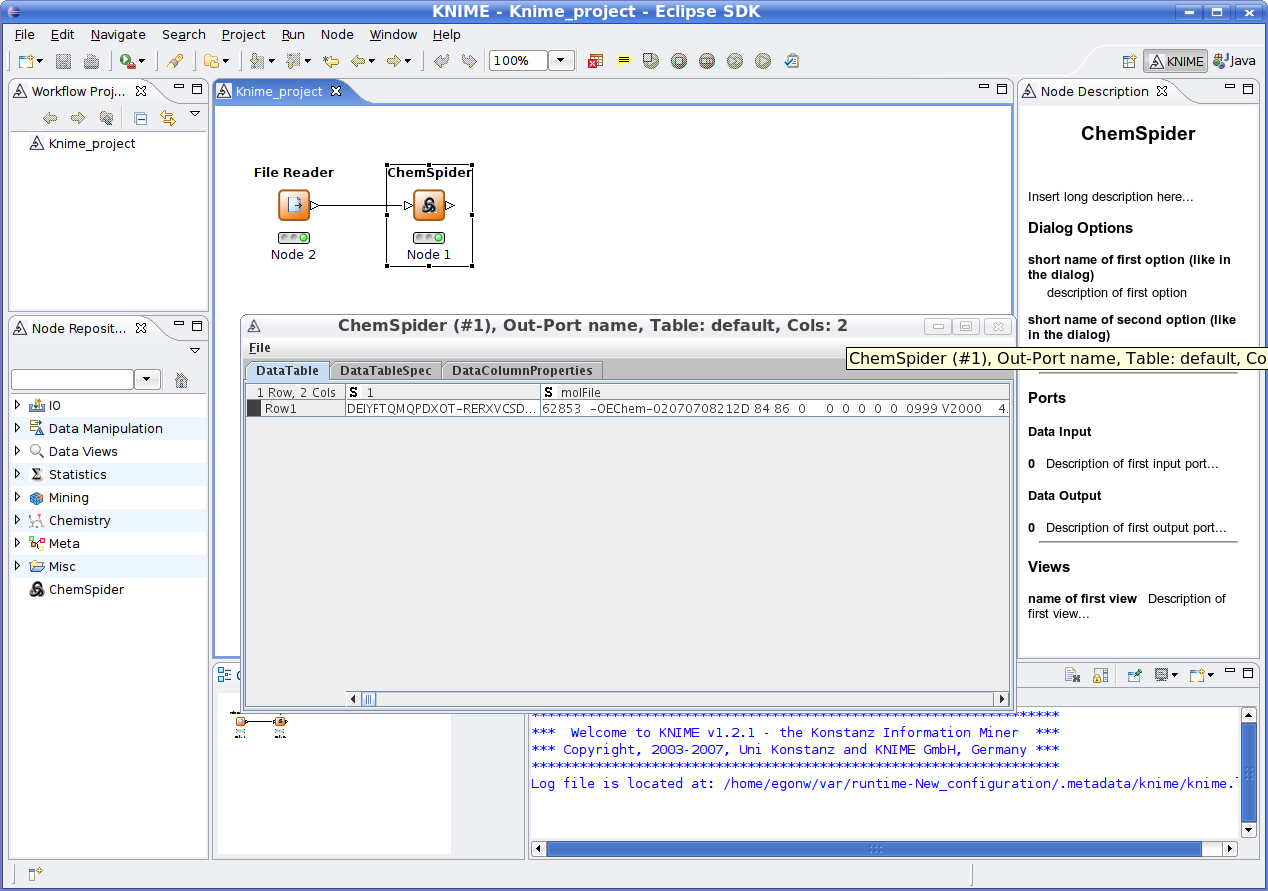
Why ChemSpider? Arbitrary. Done PubChem in the past already. Moreover, ChemSpider has the largest database of molecular structures and in that sense important to my research.
Why KNIME? Played with Taverna in the past, and expect to do much more work on Taverna in the coming year (see also this and this ). Moreover, KNIME got a CDK plugin already, and the KNIME developers contributed valuable feedback to the CDK project in the last year. It was about time that I contributed something back, though the current functionality is quite limited. KNIME has a better architectural design than Taverna1, but will face though competition with Taverna2, due next year.
The presentations
Heringa gave a presentation on network analysis, and discussed the scale-free network, hub nodes, etc, after which he gave an example on the 14-3-3 PPI family which both have promoting and inhibiting capabilities. Fraser presented work on improving microarray data analysis, by reducing non-random background noise. Schroeter presented the use of Gaussian process modeling in QSAR studies, which allows estimation of error bars (see DOI:10.1002/cmdc.200700041. I did not feel the results were very convincing, though, but the method sounds interesting. Larhlimi presented research on network analysis of metabolic networks. His approach finds so-called minimal forward direction cuts, which identifies critical parts in the network if one is interested in repressing certain metabolic processes. Hofto presented some work on the use of DFT for proteins, and picked up that one has to do things critically to be able to reproduce binding affinities. Combinations of DFT or MM with QM are becoming popular to model binding sites. Van Lenthe presented such an approach of the second day of CompLife.
By far the most interesting talk at the conference, was the insightful presentation by Paulien Hogeweg. She apparently coined the term bioinformatics. Anyway, she had a exciting presentation on feed-forward loops in relation to evolution, and showed correlation between jumps in FFL motifs with biodiversity. She also warned us for the Monster of Loch Ness syndrome, where computational models may indicate large underlying processes, which are not really existing. But that should be a problem that most of my readers should be aware of. She introduced evolutionary modeling, to put further restrictions on the models, to reduce the chance of finding monsters.
Hussong had an interesting presentation too, if one is interested in analysis of GC/MS or LC/MS data. He introduced a hard-modeling approach for proteomics data using wavelets technology. His angle on this was to use a wavelet that represents the isotopic pattern of a protein mass spectrum. Interestingly, the wavelet had negative intensities, something which one will never find in mass spectra. However, I seem to recall a mathematical restriction on wavelets that would forbid taking the squared version of the function. He indicated that the code is available via OpenMS.
Jensen, finally, presented his work at the UCC on Markov models for protein folding, where he uses the mean first passage time as observable to analyze of processes in folding state space. This allows him to compare different modeling approaches and, for example, to predict how many time steps are needed to reach folding. Being able to measure characteristics of certain modeling methods, one is able to make a objective comparison. Something which allows a fair competition .