Because of other obligations, I was unable to attend the first day of the CDK Workshop,
though Christoph had set up Skype so that at least I could hear the talks from Prof. Berthold
(Konstanz, Germany) about KNIME and Prof. Zielesny
about CDK-Taverna.
Today, Miguel Rojas and Stefan Kuhn discussed their research. Miguel showed the state of mass spectrum prediction using the CDK
and the MEDEA plugin for Bioclipse. Stefan demonstrated the NMRShiftDB
and a new lab systems for NMR experiment scheduling and management system based on that. Dr. Ott
(Nijmegen, Netherlands) showed the BioMeta Database which contains metabolite and reaction information derived from the
KEGG, but which fixes a set of chemical problems in the latter (see also the article,
DOI:10.1186/1471-2105-7-517).
The afternoons of CDK workshops traditionally have discussion sessions and hackathons. Two groups were formed: one consisted of the KNIME guys who,
together with Miguel and Federico focused in QSAR descriptor calculations in KNIME, while Stefan, Martin and me looked at the fingerprinter
peculiarities that Martin found (see also this CDK News article),
and came up with a possible further performance improvement of the AllRingsFinder. Because one class of molecules that is causing trouble consist of two
ring systems connected by a long linker, like Choloyl-CoA (below), we anticipate that splitting the molecule up into ring systems prior to using the
SSSR algorithm should speed up the complete all-ring finding process.
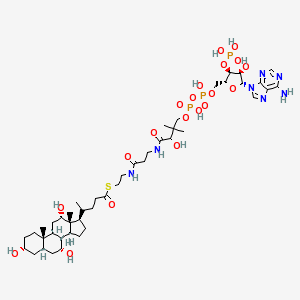
Currently, the spanning tree is calculated before deciding on using the SSSR finder, which, we think, can be used to partition the molecule
into separate ring systems. On each of them, then, the further steps of the ring search can be applied.
After dinner (pasta/pizza), during the Spanish-German handball game, we continued the hacking and discussions, now focusing as a whole group
on QSAR descriptors in KNIME. We looked at each descriptor and decided if it should go into a QSAR calculator node, or even in a node of its own.
Bugs found
I won’t close this blog entry without giving a list of problems we found in the current CDK; some minor and small, some more troublesome.
Here goes: typos all over the place; the OrderQueryBond lack a return statement in an else clause; the Mol2Reader does not mark atom and
bond aromaticity properly and reads a single bond as aromatic, and an aromatic bond as single; the Renderer2D does not always highlight
both atoms when hovering over a bond; SmilesGenerator.parseBond() should output bond orders correctly; the SSSR finder seems to have a
messed up if-else statement for the ringBondCount limit of 37; the BondCount descriptor should count all bonds by default, not just the
single bonds; IDescriptor.getParameters() should return null instead of Object[0]; several programs use the SYBYL atomtype S.o2, while
the specification and the CDK config defines S.O2; the IP descriptor now returns a variable length descriptor.
]]>
